
vir: Nobelove nagrade
Trojica je v 70. letih prejšnjega stoletja začela razvijati računalniške metode, ki omogočajo simulacije in analize velikih molekulskih sistemov. Kemiki in zlasti biokemiki so si namreč tedaj začenjali postavljati nova vprašanja. Pomembneje je postajalo, kako nek encim deluje oziroma kako nek postopek poteka, kot pa kako je posamezen encim videti v mirovanju. Podatke o strukturi encima ali drugih molekul lahko izluščimo iz rentgenske difrakcije ali jedrske magnetne resonance, a imajo pomembne omejitve. Za rentgensko difrakcijo potrebujemo kristalinični vzorec in pridobljeni rezultat so koordinate atomov v posušenem kristaliziranem encimu. Taka struktura je zelo verjetno vsaj malo drugačna kot v celicah. Za jedrsko magnetno resonanco potrebujemo vzorec v raztopini v nizki koncentraciji, od koder pridobimo podatke o medsebojni orientaciji in interakciji atomov v molekuli, a je ta še vedno v neaktivnem stanju.
Za podatke o strukturi encima med kemijsko reakcijo in napoved mehanizma reakcije nujno potrebujemo model in simulacijo, saj eksperimentalno tega ne moremo opazovati (seveda obstajajo eksperimentalne metode, ki nam pomagajo, kot je izotopsko označevanje ali femtosekundna spektroskopija).
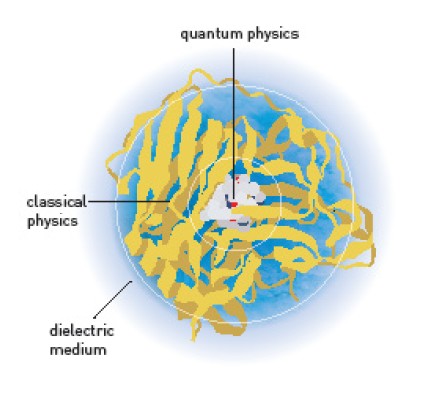
V principu lahko kemijske sisteme modeliramo na dva fundamentalno različna načina. Lahko uporabimo klasično Newtonovo mehaniko. Atomom pripišemo ustrezne polmere, naboje, interakcijske potenciale in polarizirnosti, mednje pa postavimo vezi, ki se obnašajo vzmeti. Tak model je preprost, računsko nezahteven, a ima resne omejitve. Pri spremembah molekul reaktanov v produkte je popolnoma neuporaben, pa tudi sicer zadovoljivo deluje le, če so kvantni efekti zanemarljivi.
Alternativa je modeliranje iz prvih principov, kakor imenujemo uporabo kvantne mehanike. V tem primeru rešujemo Schrödingerjevo enačbo za celoten sistem, kjer upoštevamo vsa jedra in vse elektrone. Tak sistem ima neprimerno več prostostnih stopenj, zato je računanje bistveno počasneje. Uporabimo ga lahko le za sorazmerno majhne sisteme, recimo tam do sto atomov, potem pa ne bo šlo več. Kvantna mehanika v kemiji ima sicer še druge težave, zaradi česar ni tako vsemogočna, kot bi fiziki pričakovali, a z dovolj pametno izbiro metode, baznih funkcij in potrpežljivostjo dobimo dobre rezultate - za majhne molekule.
Letošnji prejemniki Nobelove nagrade so razvili modele, ki združujejo oba pristopa. Zgodba se začne, ko je Arieh Warshel prišel na Harvard v Karplusov laboratorij. Warshel je bil strokovnjak za medmolekulske in medatomske potenciale, Karplus pa je obvladal kvantno kemijo. Warshel je na Harvard prinesel programski paket, ki sta ga v Izraelu na znamenitem Weizmannovem inštitutu v Rehovotu razvila skupaj z Levittom. Na Harvardu sta potem obdelala molekulo 1,6-difenil-1,3-5-heksatriena - torej dveh benzenovih obročev, ki sta povezana s šest ogljikovih atomov dolgo konjugirano verigo. Molekula je planarna in simetrična, zato sta uspela ločeno analizirati sigma-elektrone (to so elektroni enojnih vezi, ki so lokalizirani), ki sta jih opisala s klasično mehaniko, in pi-elektrone (to so elektroni konjugiranega sistem dvojnih vezi, ki se gibljejo po celotni molekuli). Kasneje sta pokazala, da je to mogoče v splošnem in da simetričnost molekule ni nujni pogoj.
Današnji sistemi aktivni center, kjer se vezi cepijo in nastajajo, obravnavajo kvantnokemijsko, preostali del encima klasično, medtem ko okolico, kadar je eksplicitna obravnava nemogoče, zlijejo v kontinuum. Seveda obstaja še vrsta drugih poenostavitev, kot je na primer uporaba psevdoatomov (recimo obravnava metilnih in metilenskih skupin kakor en sam atom) in podobno. Ves ta razvoj temelji na pionirskem delu, ki so ga v 70. letih začeli letošnji prejemniki Nobelove nagrade.