
Sekvenciranje s sintezo uporablja nukleotide s fluorescentnim barvilom.
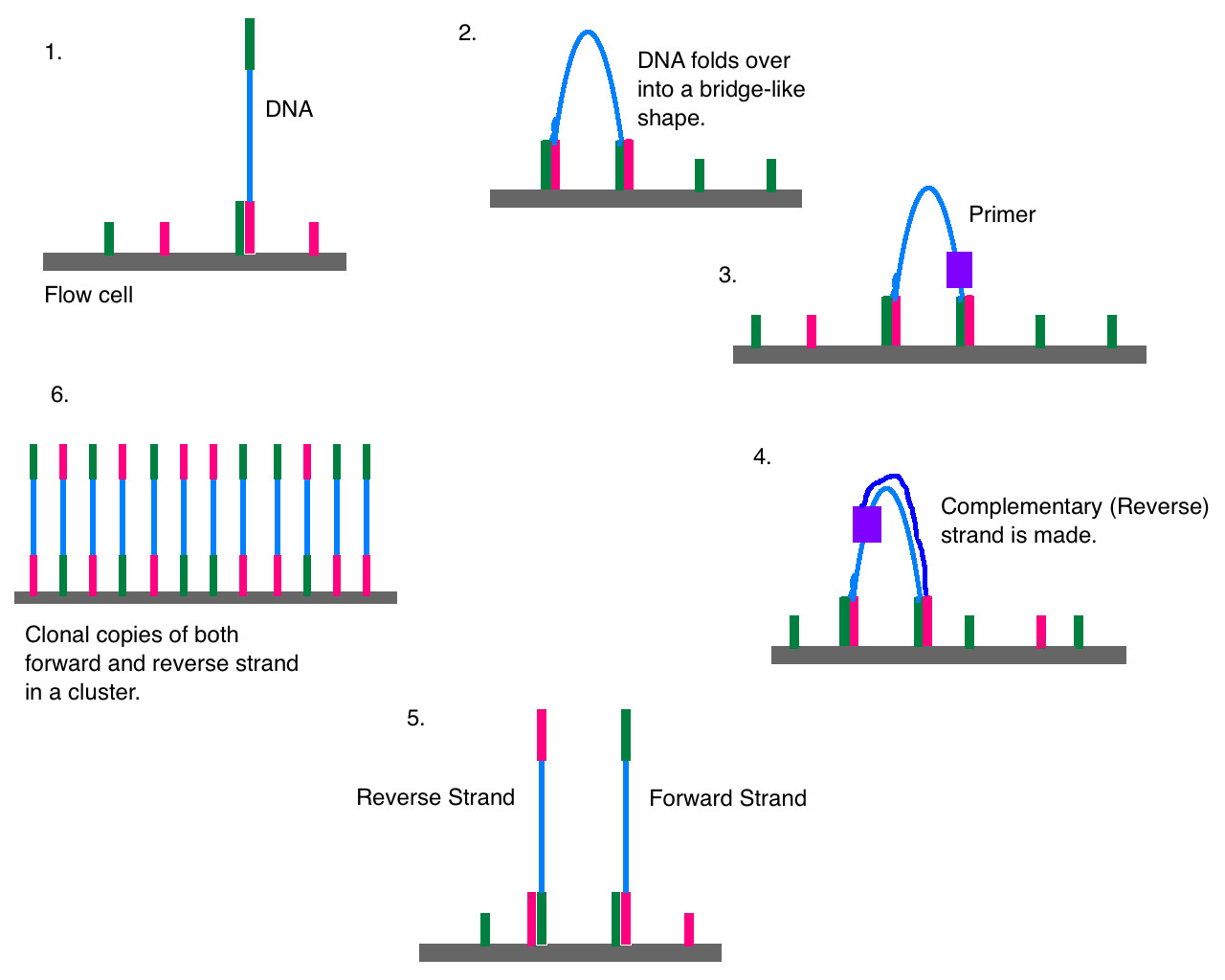
Vezava DNA na ploščo in podvojevanje z bridge-PCR.
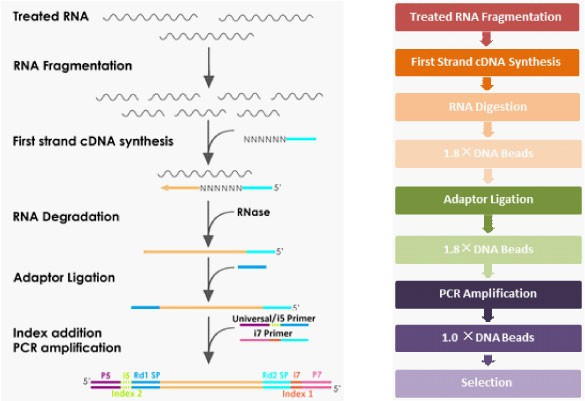
Priprava fragmentov DNA iz RNA za sekvenciranje.
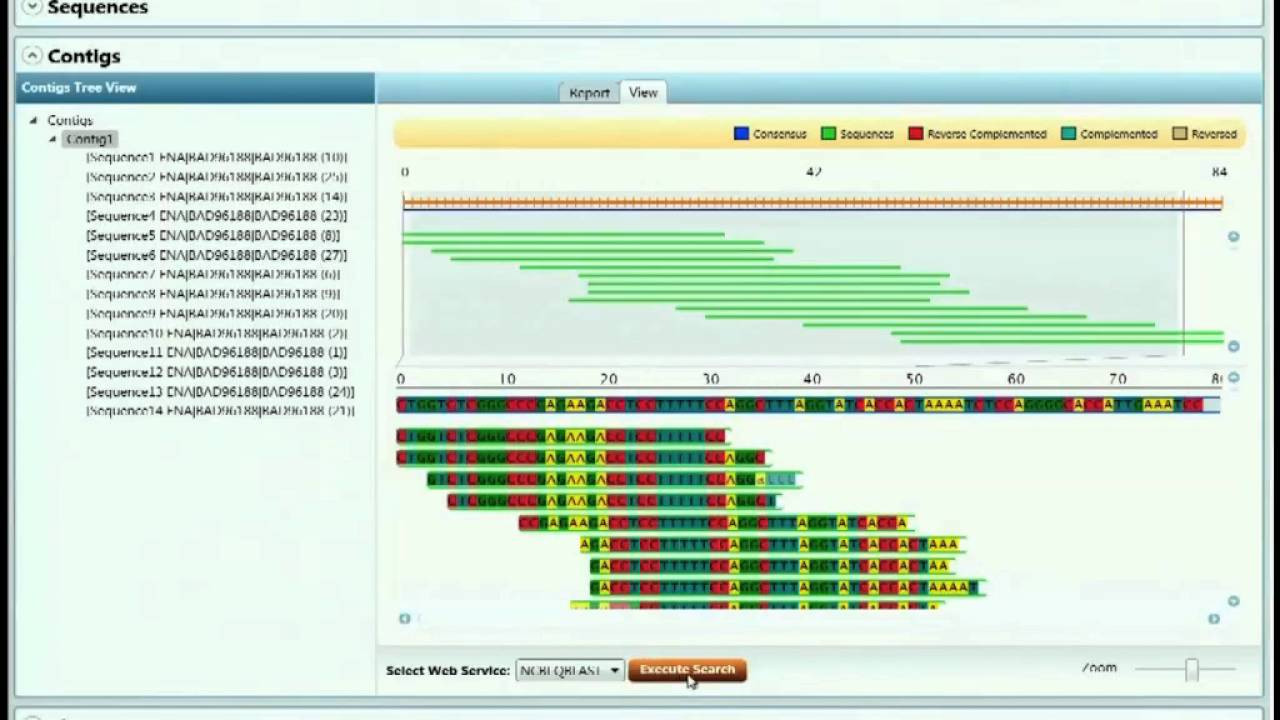
Sestavljanje končnega zaporedja iz poznanega zaporedja v fragmentih.
Primer adapterja, ki se doda na konec fragmentov DNA pred sekvenciranjem
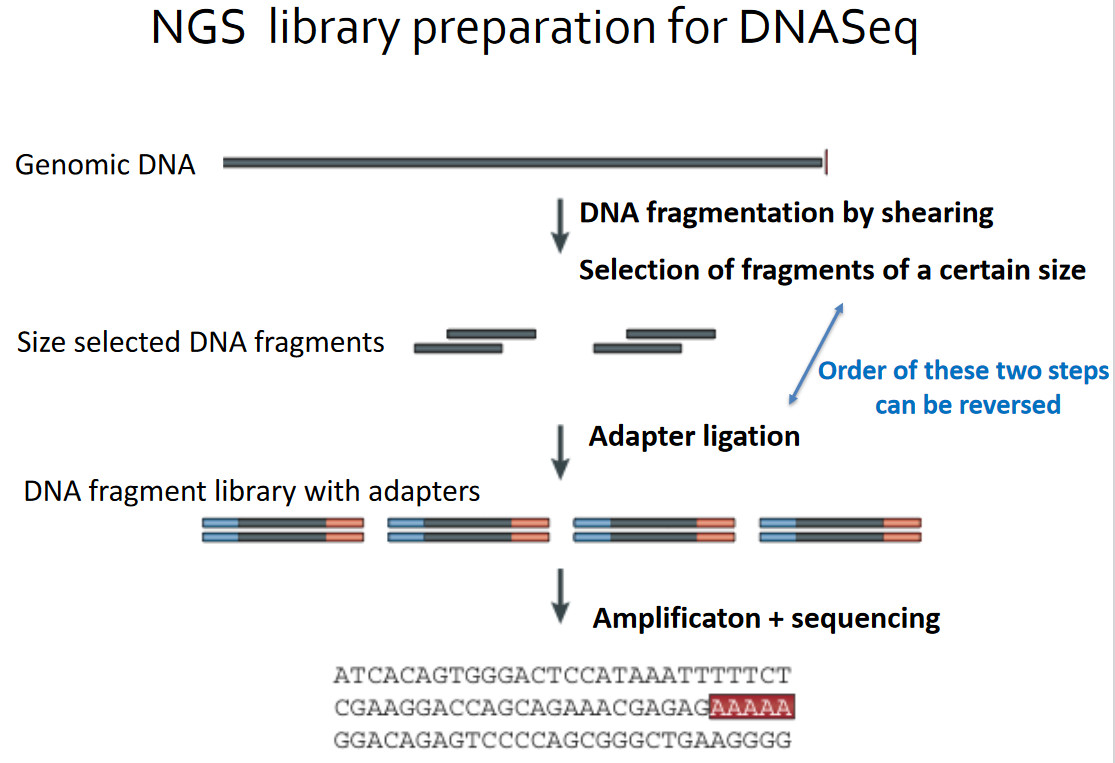
Priprava knjižnice pred sekvenciranjem.
Informacije o novem koronavirusu so Kitajci prvikrat javno objavili 31. decembra (nekaj ur pozneje tudi v angleščini), 10. januarja pa je bil celoten genom virusa že znan. Kogar zanima, si ga lahko tudi pogleda, saj je javno objavljen (originalni članek v Nature). Postopku pravimo sekvenciranje. Z njim ugotovimo, v kakšnem zaporedju so baze (adenin, gvanin, citozin in timin/uracil) v genskem zapisu. Ker imamo slovar, torej katero aminokislino posamezna trojka, poimenovana kodon, zapisuje, lahko tako hitro določimo primarno strukturo. Poglejmo, kako so določili zaporedje skoraj 30.000 baz.
Načinov za določanje zaporedja v genskem zapisu je več, odvisno od potrebe. Kadar na primer DNA uporabljamo za identifikacijo ljudi, nima nobenega smisla določevati celotnega genoma, ker smo v 99,999 % enaki - tedaj se spremljajo le regije (lokusi), ki so med ljudmi zelo variabilne. Pri nekaterih raziskavah genske zgodovine je poudarek na kromosomu Y, ki se selektivno deduje po moški liniji, ter mitohondrijski DNA, ki se selektivno deduje po materi. Spet drugače nam DNA preverijo podjetja, ki ponujajo komercialne teste DNA, saj ta gledajo večinoma dele, ki kodirajo gene, katerih funkcijo ali vsaj korelacijo poznamo. Pri koronavirusu pa je bilo treba prepoznati celoten RNA, čemur pravimo whole-genome sequencing. Marketingu navkljub tega za ljudi še nismo dosegli, čeprav se je projekt človeški genom temu zelo približal, a manjši del je še vedno nesekvenciran, predvsem pa je končni rezultat mozaik in ne genom enega posameznika. Pri koronavirusu pa so ugotovili popolnoma celoten genom, od prve do zadnje črke - kar ni nič revolucionarnega.
Samo sekvenciranje sicer ni nov postopek, saj Maxam-Gilbertovo in Sangerjevo metodo poznamo že skoraj pol stoletja, a sta počasni, zato so z njima sekvencirali zgolj viruse. Danes gre seveda drugače. Predstavljajmo si, da imamo v mikrocentrifugirki izoliran genski material. Gre za dolgo verigo sladkorjev s petimi ogljikovimi atomi (riboza v RNA ali deoksiriboza v DNA), ki so povezani preko fosfodiestrskih vezi (praktično prek fosfatnih skupin). Vsak sladkor ima pripeto bazo, ki je lahko ena izmed štirih (A, G, C, T v DNA, U v RNA). Sedaj moramo ugotoviti, v kakšnem zaporedju je nanizanih več deset tisoč baz. Zaporedje je praktično poljubno, če ne vemo nič o organizmu, od koder je material. Postopek sekvenciranja je danes že toliko izpopolnjen, da poteka skoraj avtomatično.
Šlo je takole (opis je poenostavljen, gre pa za en protokol, obstajajo različni). Vzorec so vzeli bolniku s covidom-19, in sicer iz pljuč. Vzorec je treba najprej očistiti nečistot, torej človeškega DNA in drugega RNA (denimo ribosomske RNA), ter preveriti čistost. DNA se odstrani z dodatkom DNaz, ki so encimi, ki razgrajujejo DNA. Čistost so preverili z elektroforezo, kakor se imenuje postopek, kjer izkoriščamo premikanje nabitih molekul v električnem polju. Genski material je zaradi fosfatnih skupin pri fiziološkem pH negativno nabit (zato je na primer DNA v celicah navita na pozitivne histone). Elektroforeza omogoča ločitev glede na velikost molekul.
Naslednji korak je priprava knjižnice. Postopek se razlikuje od vrste vzorca do vrste vzorca, poenostavljeno pa gre takole. Poskrbimo, da imamo le RNA, ki jo želimo sekvencirati, neželeno pa je treba odstraniti (recimo ribosome, ki so večinoma iz RNA). Iz RNA, ki je manj stabilna, se izdela prilegajoča cDNA. To zna encim reverzna transkriptaza, rezultat pa je DNA, ki ima komplementaren zapis, je pa seveda bolj stabilna od RNA - a je še vedno le enoverižna. Predstavljamo si, da smo podatke z diskete prekopirali na CD. Preostalo RNA pa je treba sedaj uničiti z RNazami (encimi, ki jo razgrajujejo). Z verižno reakcijo s polimerazo (PCR) se v prvem ciklu pridela še druga kopija DNA (to naredi encim DNA polimeraza), tako da zavzame strukturo dvojne vijačnice. V naslednjem koraku je treba DNA razrezati, saj lahko zanesljivo sekvenciramo le krajše segmente (do nekaj sto baznih parov). Nanje je nato treba pripeti adapterje - to so kratki koščki DNA, ki jih potrebujemo v postopku sekvenciranja za prepoznavo DNA. Adapterji imajo tri pomembne segmente: pritrditvenega, da se DNA sploh veže na sekvenator, indeksnega (barcode) in prijemališče za začetni oligonukleotid (primer). Rezultat je knjižnica (library), ki je pripravljena na sekvenciranje. Postopkov je več, zato poglejmo konkretno tega, ki so ga uporabili pri prvem sekvenciranju tega virusa.
To poteka na pretočni celici, ki ima ploščo, na kateri so fiksno pritrjeni komplementarni deli adapterjev. To omogoča fragmentom DNA, ki smo jim dodali adapterje, da se primejo nanje. Prvi korak je potem tako imenovani bridge PCR, ko se posamezni pritrjeni fragmenti DNA večkrat podvojijo. Ime izvira iz dejstva, da se fragment zvije v most, saj je med postopkom z obema koncema pritrjen na ploščo. Končni rezultat je več sto ali tisoč kopij istega fragmenta na plošči, kar je potrebno za zagotavljanje zanesljivosti rezultata.
Sledi ključni korak, ki je bil v konkretnem sekvenciranje s sintezo. Na plošči so fragmenti DNA v obliki enoverižne vijačnice. V celico dodajo encim DNA polimerazo (podobno kot pri PCR) in vse štiri nukleotide (dATP, dCTP, dGTP, dTTP), ki pa so ustrezno modificirani, saj imajo pripeto fluorescenčno barvilo. To ima dvojno funkcijo: blokira nadaljnjo rast verige, zato se doda le en nukleotid, hkrati pa je na vsaki vrsti nukleotida različno barvilo. Mimogrede, DNA polimeraza ne more začeti kopirati "z ničle", temveč lahko le podaljšuje verigo. Zato potrebujemo začetne nukleotide (primer), ki so vezani na začetek DNA.
Dodan je torej en nukleotid. Neporabljene nukleotide sperejo, sekvenator pa "posname fotografijo" plošče, s čimer iz barve fluorescirajočega barvila dobimo informacijo, kateri nukleotid se je pripel kam na ploščo. Nato se s spiranjem odstrani fluorescirajoči del, s čimer postane nastajajoča veriga DNA spet pripravljena na rast. V naslednjem koraku se spet lahko pritrdi le en nukleotid, spet se posname stanje na plošči in tako dobimo informacijo o drugem nukleotidu. Postopek ponavljamo do dolžine fragmenta. Ker je na plošči pritrjeno veliko fragmentov, ki so vsak na svojem, točno določenem mestu, poteka sekvenciranje paralelno. Kaj pa sedaj?
Pomembno vlogo ima bioinformatika (več informacij za programerje). Pri takšnem sekvenciranju SARS-CoV-2 so na primer dobili 56 milijonov odčitkov različnih dolžin (okrog 150 baznih parov). To si lahko predstavljamo, kot če bi imeli milijone kopij knjige (RNA celotnega virusa) brez oštevilčenih strani, potem pa bi vsako raztrgali na različno dolge odseke in iz njih želeli obnoviti originalno besedilo. Z računalnikom je treba zložiti te strani nazaj, pri čemer si pomagajo s prekrivanjem zaporedij na koncih fragmentov. Upoštevati je treba tudi, da nekateri odčitki ne sodijo zraven, saj ustrezajo ostankom tujega genskega materiala (človeški DNA) ali adapterjem, ki so se uporabljali v postopku. Računalniške metode analizirajo milijone fragmentov, polovijo morebitne napake pri posameznih nukleotidih, in izpljunejo končno zaporedje. S PCR se preveri, da je to zaporedje res pravilno, in določi še nukleotide na koncih (genome termini). In tako smo dobili genski zapis, ki je bil izhodišče ne le za pripravo cepiva, temveč študije provenience virusa, njegovega delovanja in tudi - mutacij.
Določevanje genoma svežih vzorcev virusa namreč še vedno poteka, načinov pa je več (kar opisuje ECDC). Ne določa se sicer pri čisto vsakem vzorcu, a dovolj pogosto, da sčasoma najdejo tudi mutacije, kot je britanska. Le tako namreč lahko spremljamo širjenje in spreminjanje sevov - že sedaj je jasno, da sev, ki trenutno kroži po Evropi, ni enak izvornemu.