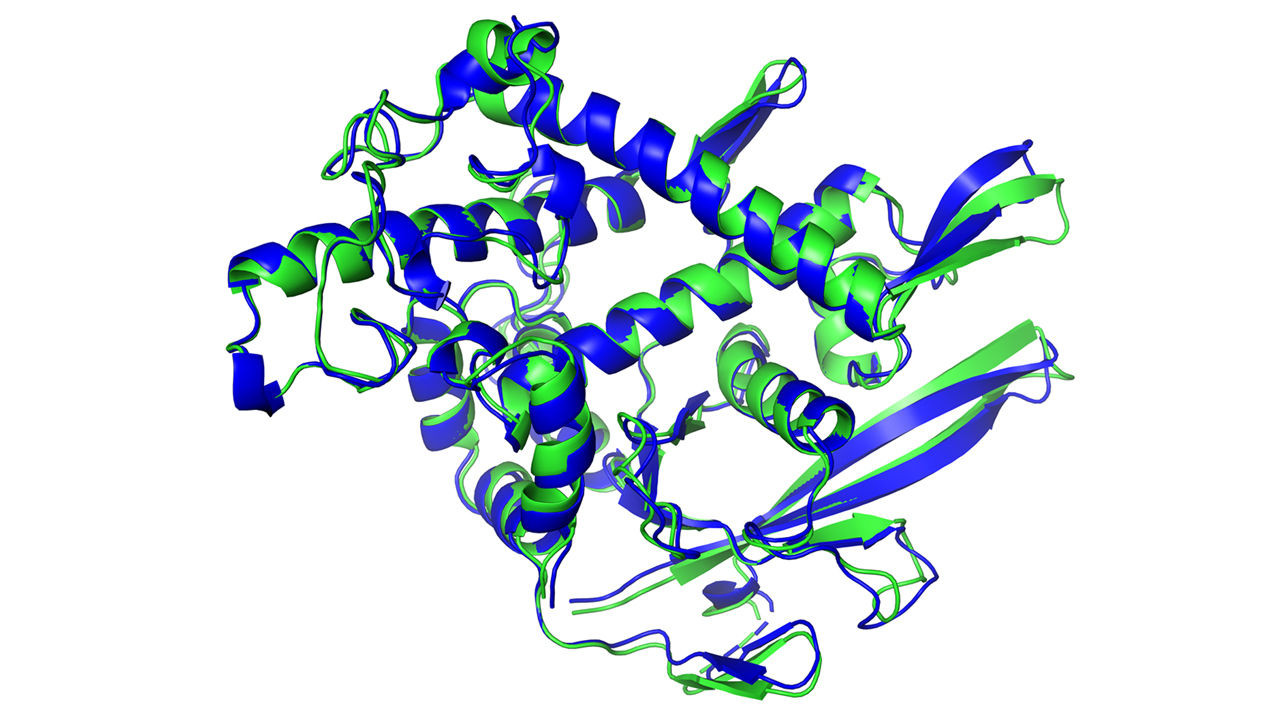
zgradba beljakovine, pri čemer je modro označena AlphaFoldova ocena, zeleno pa mikroskopska meritev
vir: DeepMindBeljakovine so funkcijsko najbolj raznolika skupina molekul v živih organizmih, saj skrbijo za vse od katalize reakcij do rokovanja z dednino. Kaj natančno neka beljakovina počne, je odvisno tako od njene kemijske sestave kot tudi njene natančne prostorske strukture - se pravi, kako se njeni elementi spontano razporedijo, oziroma, kako se beljakovina "zvije". V osnovi skušamo natančno zgradbo dognati z opazovalnimi metodami, kot sta rentgenska difrakcija in krioelektronska mikroskopija. A te imajo omejitve, saj v mnogih primerih vzamejo dosti časa in imajo resne probleme z nekaterimi sortami proteinov, na primer onimi, ki so vpeti v druge strukture, kot so celične membrane. V začetku sedemdesetih let prejšnjega stoletja je nobelovec Christian Anfinsen pričel popularizirati idejo, da je zvitje beljakovin odvisno zgolj od njihove kemijske sestave, oziroma sekvence aminokislin, ki jih gradijo. Tako se je rodilo področje analize zvijanja proteinov z računalnikom, s katero smo že pred desetletji znali dognati strukturo najbolj preprostih med njimi. Toda pri kompleksnejših - kamor sodi večina uporabnih - smo napredovali po polžje, kajti računsko gre za neznansko zahteven izziv, če se ga lotimo preprosto z pregledovanjem vseh možnosti, kako se večja molekula lahko zvije. To nam nakazuje že dejstvo, da je stroka v ta namen uporabila metode porazdeljenega računalništva, med katerimi je brez dvoma najbolj znan projekt Folding@home.
Da bi področju dali nekaj dodatnega zaleta, so leta 1994 zasnovali tekmovanje CASP ali Critical Assessment of Structure Prediction, ki poteka na dve leti. Tekmovalnim skupinam vsakič predložijo okoli sto proteinov, katerih zgradba je bila ravno dognana z opazovalnimi metodami in torej še ni javna. (Doslej smo opredelili strukturo zgolj 170.000 beljakovin od približno 200 milijonov odkritih, oziroma milijard teoretično obstoječih.) Uspeh na CASPu se preudarja glede na to, kako blizu je izračunana ocena dejanski zgradbi, rezultati pa se beležijo v lestvico od 1 do 100, pri čemer vrednost nad 90 pomeni, da je ocena tako blizu realnosti, da je razkorak že lahko posledica merske napake. Dolga leta so bili rezultati za vse malo bolj zapletene proteine porazni, med 20 in 40. Svež veter pa je zavel pred dvema letoma, ko se je izziva prvič lotil DeepMindov algoritem AlphaFold in že "iz prve" pometel s tekmeci. Toda absolutni izkupiček - nekaj nad 60, še ni bil dovolj, da bi bil uspeh tudi praktično uporaben. Strokovnjaki so menili, da nas od tega loči vsaj še desetletje. DeepMindu je uspelo v dveh letih.
Algoritem AlphaFold 2 je pri dveh tretjinah beljakovin na letošnjem tekmovanju dosegel oceno nad 90, pri večini preostalih pa okoli 85; izjema je bil poseben proteinski kompleks, kakršni mu očitno še delajo probleme in bodo naslednji fokus DeepMindovih inženirjev. V praksi to pomeni, da so zgradbo večine beljakovin zadeli na približno poldrugi angstrem natančno, se pravi za velikost atoma. Podrobnejšo sestavo algoritma bodo predstavili na konferenci, ki te dni poteka ob robu predstavitve rezultatov tekmovanja. Vemo, da obstaja kar nekaj razlik glede na prvo generacijo AlphaFolda. Takrat je bilo globoki nevronski mreži dodeljeno le "ugibanje" o interakciji med pari aminokislin, medtem ko strojno učenje sedaj vodi ves postopek, poleg pa so dodali tudi nekaj trdno vprogramiranih fizikalnih omejitev. Kar se tiče same nevronske mreže, so uporabili moderno podvrsto globokega učenja, "attention network", ki se zna osredotočati na manjše odseke in protein "graditi kot sestavljanko". Urili so jo na bazi vseh doslej poznanih molekul, in to z razmeroma pohlevnim superračunalnikom z zgolj 128 procesorji.
DeepMind je v zadnjih letih odločno zakorakal s področja iger, kjer je strašil z algoritmom AlphaGo, v znanstveno in medicinsko sfero, za štart predvsem v diagnostiko. Toda uspeh AlphaFolda 2 je prebojen na bazični ravni in bo imel zaradi tega po mnenju množice strokovnjakov veliko bolj daljnosežne posledice. Ne gre le za teoretični začetek rešitve "petdesetletnega problema" v biokemiji, temveč za takojšen bistveni premik v naši sposobnosti analiziranja beljakovin, saj je AlphaFold 2 v preteklem mesecu že pomagal razkriti strukture molekul, ki so dotlej raziskovalce begale desetletja. To je ključno za delovanje zdravil, prav lahko pa pomeni tudi preporod področja izdelave sintetičnih proteinov. Zgolj okoli četrtina od 20.000 različnih beljakovin v človeškem telesu je namreč natančno poznana, zato je prostora za napredek v farmaciji še ogromno. Poleg težav s proteinskimi kompleksi je še ena šibka točka DeepMindovega algoritma trenutno počasnost, saj za analizo posamezne molekule potrebuje več dni; zato še vedno ostaja možnost uporabe malo manj natančnih, a hitrejših rešitev. Vprašanje je tudi, kako se spopada z drugimi eksotičnimi molekulami, ki funkcionirajo spojene z drugimi elementi. V DeepMindu pravijo, da bodo pri svoji razlagi programja dovolj podrobni, da bo imela od tega korist vsa skupnost; v začetku prihodnjega leta sledi tudi znanstveni članek.